Hu Hu1,
Ming-Jun Dong2,3,
Jun Zhang3 ![]()
For correspondence:- Jun Zhang Email: zhangj310013@gmail.com Tel:+8657187964033
Received: 6 July 2015 Accepted: 17 December 2015 Published: 28 February 2016
Citation: Hu H, Dong M, Zhang J. A holistic in silico approach to develop novel inhibitors targeting ErbB1 and ErbB2 kinases. Trop J Pharm Res 2016; 15(2):231-239 doi: 10.4314/tjpr.v15i2.3
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To design a dual inhibitor of natural origin capable of targeting ErbB1 and ErbB2 kinases for the treatment of lung cancer.
Methods: Advanced In silico drug designing techniques were explored in this study. Sequence and structure analysis of ErbB1 and ErbB2 was followed by three dimensional (3D) pharmacophore. The generated model was used for molecular docking simulation studies for predicting the best natural dual inhibitors, the selected inhibitors were subjected to absorption, distribution, metabolism, excretion and toxicology (ADME/Tox) prediction.
Results: The results confirmedfive phytochemicals, viz. hyoscyamide, cannabisin F, cochinchinenene D, cannabisin E, and heliotropamide and five FDA approved drugs namely fesoterodine, antrafenine, fluspirilene, posaconazole, and iloprost to be potential inhibitors of both ErbB1 and ErbB2. The shortlisted compounds from both the panels were showing better MolDock score than the two reference drugs (Lapatinib and Afatinib).
Conclusion: The 3D pharmacophore modelling and molecular docking simulations gave us ten compounds that successfully exploited dual inhibition of ErbB1 and ErbB2. With 8 and 12 hydrogen bonds with ErbB1 and ErbB2 respectively cannabisin F showed best interaction of all.
Introduction
The incidence of lung cancer worldwide is 13 %. The highest incidence is in developed countries, particularly in North America and Europe, and is less common in developing and underdeveloped countries [1-3]. The etiology of this malady is linked with the anomaly in around 26 genes, among them receptor tyrosine kinases (RTKs), are involved in lung cancer proliferation [4,5]. RTKs belong to epidermal growth factor receptor family and there four structurally related family members are ErbB1 (also known as EGFR or Her1), ErbB2 (also known as Her2 or Neu), ErbB3 (also known as Her3), and ErbB4 (also known as Her4). Among these, ErbB1 and ErbB2 are predominant in lung cancer [2,6-8]. These kinases have an extracellular ligand binding domain, a single hydrophobic transmembrane domain, and an intracellular tyrosine kinase domain [9]. The success rate of antibody treatment targeting the extracellular ligand binding domain is low [6]. The tyrosine kinase domain, which is structurally conserved in both ErbB1 and ErbB2, is the target site for most of the successful anti-cancer drugs which block its kinase activity. Many previous studies have adapted a dual targeting approach—inhibiting ErbB1 and ErbB2 concurrently–which provides synergistic inhibition [10,11]. The current study also follows the same approach, but the difference being that we focus on single inhibitor targeting both proteins, unlike the previous approaches which have considered individual inhibitors for each protein.
Methods
Protein preparation
The primary sequences of ErbB1 (UniProt ID: P00533) and ErbB2 (UniProt ID: P04626) were retrieved from Universal Protein Resource (UniProt), which is a comprehensive resource for protein sequence and annotation data. The three-dimensional structures of ErbB1 (PDB ID: 4G5J) and ErbB2 (PDB ID: 3PP0) were obtained from Protein Data Bank (PDB). Although ErbB1 has four isoforms, only the first isoform is primarily involved in lung cancer. ErbB2 has five isoforms. To understand the similarities and dissimilarities of the kinase domain of these isoforms, sequence comparison was performed for each protein separately using the Clustal Omega program of Uniprot. The three-dimensional structures of both proteins were compared by jCE algorithm of RCSB PDB Protein Comparison Tool.
Natural product library construction
Natural products containing 6,160 compounds from marine sources and 18,151 compounds from medicinal plants were retrieved from previous extensive literature studies [12,13], DR. Duke’s Phytochemical Library (http://www.ars-grin.gov/duke/), and PubChem BioAssay (http://www.ncbi.nlm.nih.gov/pcassay), which contains the bioactivity screens of chemical substances. Reference approved drug molecules Afatinib (DrugBank ID: DB08916) and Lapatinib (DrugBank ID: DB01259), as well as the 1,543 molecules of FDA approved drugs were obtained from DrugBank (www.drugbank.ca). Since the approved drugs were selected because of their FDA approval, they did not require pre-clinical trials. DrugBank is a comprehensive, high-quality, freely accessible, online database containing information on drugs and drug targets.
Ligand preparation and ADME/Tox prediction
The ligands used for the docking studies were prepared using Maestro version 9.0. It is a robust collection of tools designed to prepare high quality, all-atom 3D structures for large numbers of drug-like molecules, starting with 2D or 3D structures in SD or Maestro format. Hydrogens were added to the ligands. The ligand were desalted and tautomers were generated. Specified chiralities of the ligand were maintained. Low energy ring conformation was generated from each ligand. The geometry was optimized using forcefield OPLS 2005. The output file was generated in the sdf file format. QikProp was performed for the natural product molecules obtained from LigPrep. This application was used to determine the Absorption, Distribution, Metabolism, and Excretion (ADME) property. Thirty-one QikProp parameters were considered for each molecule.
It is widely known that QikProp can efficiently evaluate pharmaceutically relevant properties for over half a million compounds in one hour, making it an indispensable lead generation and lead optimization tool. The descriptors considered were number of rotatable bonds, molecular weight, computed dipole moment, total solvent accessible surface area, total solvent-accessible volume, number of hydrogen bond donor, number of hydrogen bond acceptor, number of likely metabolic reactions, van der Waals surface area of polar nitrogen and oxygen atoms and carbonyl carbon atoms, predicted central nervous system activity, hexadecane/gas partition coefficient, octanol/gas partition coefficient, water/gas partition coefficient, octanol/water partition coefficient, aqueous solubility, IC50 value for blockage of HERG K+ channels, apparent Caco-2 cell permeability in nm/sec, apparent MDCK cell permeability in nm/sec, brain/blood partition coefficient, binding to human serum albumin human oral absorption on 0 to 100 % scale, and conformation-independent predicted aqueous solubility. The molecules that passed ADME prediction were used for Toxpredict procedures. Toxpredict (http://apps.ideaconsult.net:8080/ToxPredict) is a tool from the OpenTox server which calculated the toxicity of the natural product molecules.
3D pharmacophore modelling and virtual screening
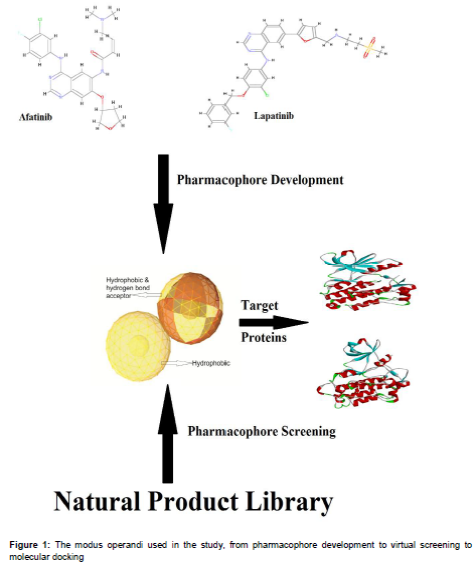
A pharmacophore is defined as a 3D structural feature that illustrates the interaction of a ligand molecule with a target receptor in a specific binding site. It is possible to compute the shared pharmacophore feature of a known drug when its three-dimensional structure is available. To this end, virtual screening of the ligand based 3D pharmacophore was performed using LigandScout. The reference drug molecules Afatinib (DrugBank ID: DB08916) and Lapatinib (DrugBank ID: DB01259) were optimized by LigPrep of Maestro version 9.0. Both the optimized structures were loaded onto the LigandScout, both structures were then aligned based on the pharmacophore features. Virtual screening was then performed with the generated shared feature against the natural products database and approved drug database created from the molecules obtained from the previous step which are passed from the in silico ADME prediction. The “pharmacophore-fit” scoring function, “match all query features” screening mode, and “get best matching conformation” retrieval mode were used for the pharmacophore search. The screened molecules were further confirmed by docking study.
Molecular docking
Molecular docking studies were carried out for both the proteins separately with both the datasets natural product and approved drugs using Molegro Virtual Docker (MVD). It has two docking search algorithms: MolDock Optimizer and MolDock Simplex Evolution (SE). MolDock Optimizer is the default search algorithm in MVD. In order to dock the receptor and ligand, the receptor was prepared from the “prepare molecule” option provided. Then, for grid searching, cavities were generated using the “detect cavity” option. Finally, the ligands obtained from the pharmacophore studies were provided in an sdf file format for docking using the docking wizard. Default parameters with the grid resolution of 0.3 A were employed for elucidating single molecules, capable of inhibiting both the kinases.
Visualization of results
Molegro Virtual Docker (MVD) software, which is an integrated platform for predicting protein–ligand interactions, was used to visualize the docked result. This software handles all aspects of the docking process from preparation of the molecules to determination of the potential binding sites of the target protein, and prediction of the binding modes of the ligands.
Results
Protein preparation
The retrieved isoform sequences of ErbB1 and ErbB2 were aligned independently. The kinase domain of isoform1 of ErbB1 did not show any significant similarities with that of the other three isoforms. The kinase domains of all the five isoforms of ErbB2 show significant similarities. The three-dimensional structures of both proteins were compared by the jCE algorithm. The kinase domains of both proteins showed higher similarity with RMSD: 1.68, identities: 73 % and similarities: 83 %, indicating that a common inhibitor may be inhibiting both the proteins.
ADME/Tox prediction
It was determined from ADME/Tox prediction that from 24,311 natural product molecules only 1,015 screened out were more poised. The significant reduction in the number of molecules could be attributed to the many stringent descriptors considered for this prediction.
3D pharmacophore modelling and virtual screening
The structurally optimized drug molecules, Afatinib and Lapatinib, were aligned based on their pharmacophore features using LigandScout. Three shared features - two hydrophobic and one hydrogen bond acceptor - were obtained from both the drugs (), which were considered as a query for virtual screening.
The 3D pharmacophore virtual screening demonstrated that among the 1,015 ADME passed natural product molecules, only 201 molecules have the pharmacophore features of both drugs, and from the 1527 approved drugs, 356 molecules were matched with the query features. The ensued molecules were further confirmed by docking study.
Molecular docking and toxicity prediction
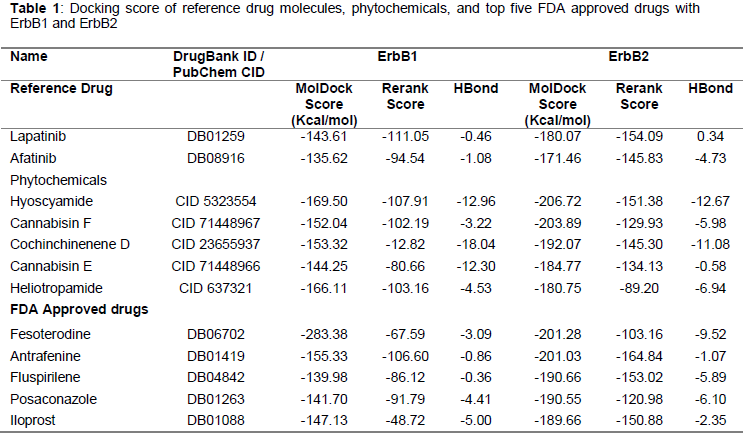
The docking studies demonstrated that only 109 of the 201 natural product molecules and 125 of the 356 approved drugs have efficient binding with both the targets. Furthermore, only eighteen natural product molecules and five approved drugs were found to have a better docking score than the reference drug molecules. The two-dimensional structure of the reference drug molecules, five best nontoxic natural product molecules and top five FDA approved drug molecules are shown in Figs 2(a) and 2(b).
The MVD docking score is shown in . The selected phytochemicals had a docking score ranging from −144.25 Kcal/mol to -206.72 Kcal/mol and the top ranked approved drugs had a docking score ranging from −139.98 Kcal/mol to −201.28 Kcal/mol, which is better than that of the reference drug molecules.
The docking result with hydrogen bond interactions are shown in Figs. 3(a) and 3(b). The number of hydrogen bonds and the interacting residues are shown in Tables 2 and 3.
The reference drug molecules had one to four hydrogen bond interactions with the target. More importantly, the shortlisted phytochemicals and FDA approved drugs had six to twelve and one to eight hydrogen bond interactions, respectively. The docking score and number of hydrogen bonds of natural products and FDA approved drugs demonstrated higher affinity with proteins than the reference drug molecules. Furthermore, a detailed analysis of the docking results showed that all ligands bonded with the kinase domains of the target.
Discussion
Molecular docking has played a key role in the identification of efficient binding of receptors and ligands. Compounds identified from docking studies with most favourable binding energy were considered as hits. Hyoscyamide, a secondary metabolite present in the seeds of Hyoscyamus niger is binding to ErbB1 and ErbB2 with MolDock score of -169.5 (kcal/mol) and -206.7 (kcal/mol) respectively. Hyoscyamide binding energy is the least calculated among the top ten compounds with ErbB2. The hydrogen bond forming pattern is nine hydrogen bonds with ErbB1 and seven with ErbB2. Cannabisin F and Cannabisin E are acyclic bis-phenylpropane lignanamides existing in the fruits of Cannabis sativa. Cannabis sativa is cultivated for seed oil, food, and medicine. Historically, tinctures, tea, and ointments have also been common preparations. In traditional Indian medicine, C. sativa has been used as a hallucinogenic, hypnotic, sedative, an analgesic, and anti-inflammatory agent [14]. Cannabisin E is binding to ErbB1 and ErbB2 with MolDock score of -144.2 (kcal/mol) and -184.7 (kcal/mol), the hydrogen bond pattern with the two kinases is seven and eleven respectively. Cannabisin F is the best compound among shortlisted phytochemicals in terms of hydrogen bond pattern. Cannabisin F is forming eight hydrogen bonds with ErbB1 and twelve with ErbB2. Recently, a group of scientists from China synthesized cannabisin F from vanillin [15]. Cannabisin F has already been reported for its cytotoxic activity [16], and cell-growth inhibitory activities against human lung cancer and human cervical cancer [17]. Cochinchinenene D from Dracaena cochinchinensis has already been reported for its antibacterial activities against Helicobacter pylori [18], Dracaena cochinchinensis is predominant in China and its various other secondary metabolites are used to cure a variety of diseases, including neurodegenerative diseases, diabetes, and cancer [19,20]. Cochinchinenene D is forming the best interaction with ErbB1of ten hydrogen bonds, which is the best among the analyzed twelve compounds. Heliotropamide from Heliotropium ovalifolium has oxopyrrolidine-3-carboxamide central moiety. Heliotropamide among the selected phytochemicals is showing the least number of interaction with ErbB1 and ErbB2 (seven and six).
The hydrogen bond binding pattern of the FDA-approved drugs is overall lower than phytochemicals. Fesoterodine, which is an approved drug, a prodrug is showing the best binding energy of -283.3 (kcal/mol) with ErbB1. The compound have no reported anti-cancer activity, however its in vivo broken down active metabolite: 5-hydroxymethyl tolterodine (5-HMT) exhibits an antimuscarinic activity by acting as a competitive muscarinic receptor antagonist, Both urinary bladder contraction and salivation are mediated via cholinergic muscarinic receptors [21]. Antrafenine is a piperazine derivative drug that acts as an analgesic and anti-inflammatory with an efficacy similar to that of naproxen, the compound is showing second best binding energy with ErbB2 among FDA-approved drugs and has not been reported for anti-cancer activity. Fluspirilene is a relatively long-acting injectable depot antipsychotic drug used for schizophrenia. It does not differ greatly from other depot antipsychotics (fluphenazine decanoate, fluphenazine enathate, perphenazine onanthat, pipotiazine undecylenate) with respect to treatment efficacy, response, or tolerability. Fluspirilene is showing the least potential for inhibiting the selected kinases among the ten shortlisted compounds. Posaconazole among the FDA-approved drugs is showing the best hydrogen bond interaction with both ErbB1 and ErbB2. The compound exerts its antifungal activity through blockage of the cytochrome P-450 dependent enzyme, sterol 14α-demethylase, in fungi by binding to the heme cofactor located on the enzyme, this leads to the inhibition of the synthesis of ergosterol, a key component of the fungal cell membrane, and accumulation of methylated sterol precursors, which in turn results in the inhibition of fungal cell growth, and ultimately, cell death [22]. Iloprost is a second generation structural analog of prostacyclin (PGI) with about tenfold greater potency than the first generation stable analogs, such as carbaprostacyclin. The compound is forming two and four hydrogen bonds with ErbB1 and ErbB2 respectively, which is second best interaction among FDA-approved drugs.
Conclusion
The in silico approach has helped us to predict promising compounds for targeting ErbB1 and ErbB2 kinases with single inhibitor for the effective treatment of lung cancer. The approach provides an opportunity to focus only on selected compounds for further wet-lab experimental analysis.
The generated data of the ten compounds show immense potential in targeting both kinases with single inhibitor. All the selected lead compounds are non-toxicity, the factor that has an immense role in drug discoveries.
Declarations
Acknowledgement
References
Archives


News Updates